Development of New Drug-Like Molecules to Treat Cystic Fibrosis: Cystic fibrosis (CF) is a devastating genetic disease with those afflicted possessing an average life expectancy of only 40 years. CF is characterized by thick mucus in the lungs and eventual respiratory failure. Most of the available treatments for CF focus only on the symptoms of the disease (chronic bacterial lung infections); however recent breakthroughs have discovered the first drugs (Ivacaftor and Orkambi) that treat the underlying cause of CF.
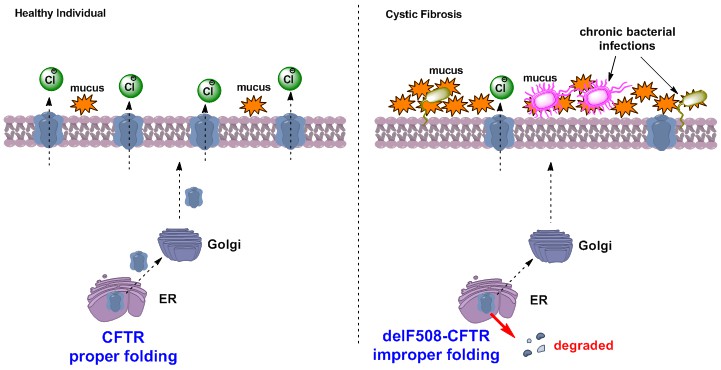
CF is caused by mutations to the cystic fibrosis transmembrane conductance regulator (CFTR) ion channel. Located in epithelial cells of the lungs and other organs, CFTR mediates chloride secretion. Deletion of phenylalanine 508 (delF508-CFTR) is the most prominent CFTR mutation, occurring in greater than 70% of CF patients. delF508-CFTR exhibits impaired protein folding, leading degradation of the mutated protein and low levels of expression of expression in the lungs. As a result, delF508-CFTR mediated chloride secretion is ~3% of that observed in non-CF lung epithelia. Low chloride secretion is associated with the build-up of thick mucus on the lungs and this mucus harbors harmful bacteria. Over time these chronic bacterial infections lead to lung failure and death.
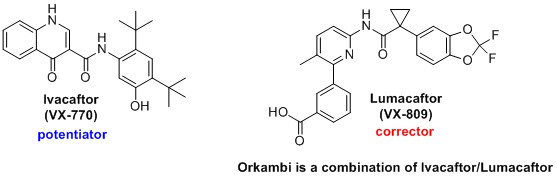
Small molecule modulators of delF508-CFTR are a promising strategy to combat CF. These compounds are designed to correct the folding deficiency (i.e. “correctors”) and/or increase chloride transport (i.e. “potentiators”). In collaboration with Professor Stephen Aller (University of Alabama, Birmingham), our lab is working to discover new corrector and potentiator compounds to help combat CF.